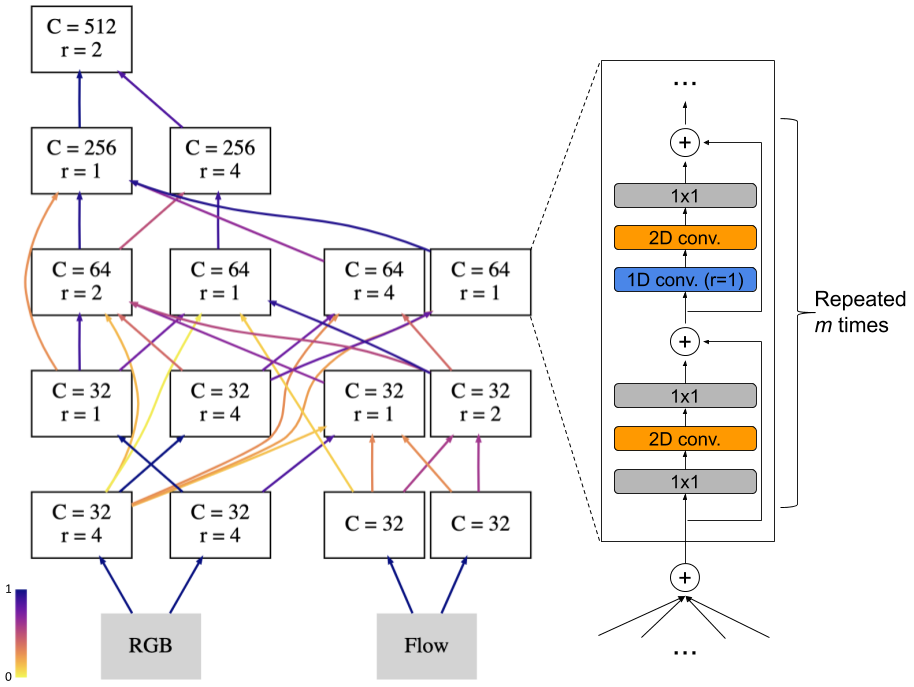
Today we are open-sourcing Magika, Google’s AI-powered file-type identification system, to help others accurately detect binary and textual file types. Under the hood, Magika employs a custom, highly optimized deep-learning model, enabling precise file identification within milliseconds, even when running on a CPU.
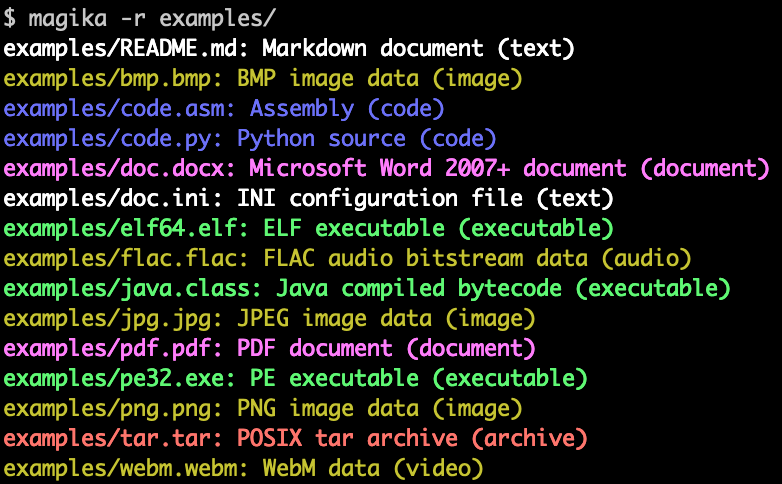 |
| Magika command line tool used to identify the type of a diverse set of files |
You can try the Magika web demo today, or install it as a Python library and standalone command line tool (output is showcased above) by using the standard command line pip install magika.
Why identifying file type is difficult
Since the early days of computing, accurately detecting file types has been crucial in determining how to process files. Linux comes equipped with libmagic and the file utility, which have served as the de facto standard for file type identification for over 50 years. Today web browsers, code editors, and countless other software rely on file-type detection to decide how to properly render a file. For example, modern code editors use file-type detection to choose which syntax coloring scheme to use as the developer starts typing in a new file.
Accurate file-type detection is a notoriously difficult problem because each file format has a different structure, or no structure at all. This is particularly challenging for textual formats and programming languages as they have very similar constructs. So far, libmagic and most other file-type-identification software have been relying on a handcrafted collection of heuristics and custom rules to detect each file format.
This manual approach is both time consuming and error prone as it is hard for humans to create generalized rules by hand. In particular for security applications, creating dependable detection is especially challenging as attackers are constantly attempting to confuse detection with adversarially-crafted payloads.
To address this issue and provide fast and accurate file-type detection we researched and developed Magika, a new AI powered file type detector. Under the hood, Magika uses a custom, highly optimized deep-learning model designed and trained using Keras that only weighs about 1MB. At inference time Magika uses Onnx as an inference engine to ensure files are identified in a matter of milliseconds, almost as fast as a non-AI tool even on CPU.
Magika Performance
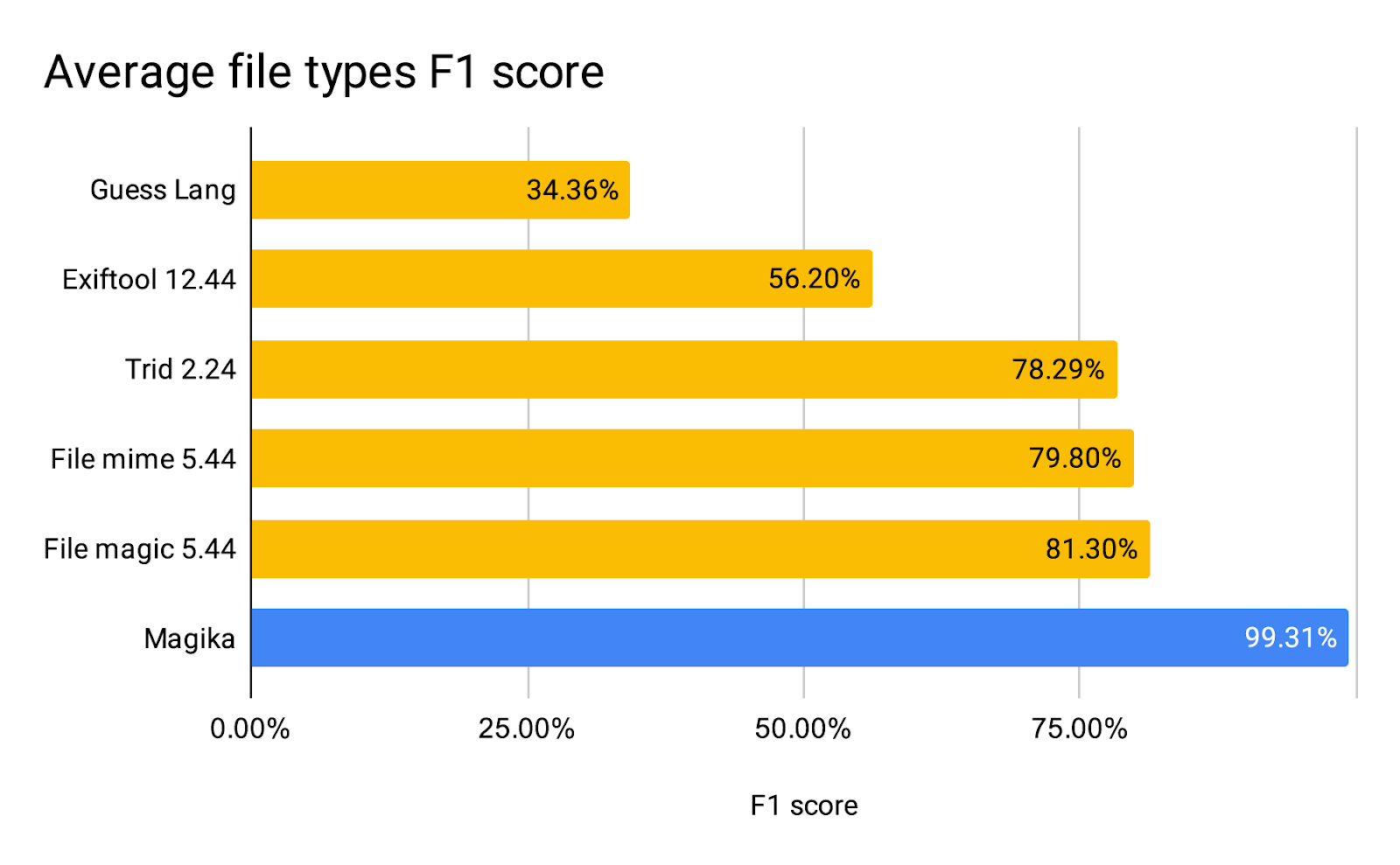 |
| Magika detection quality compared to other tools on our 1M files benchmark |
Performance wise, Magika, thanks to its AI model and large training dataset, is able to outperform other existing tools by about 20% when evaluated on a 1M files benchmark that encompasses over 100 file types. Breaking down by file type, as reported in the table below, we see even greater performance gains on textual files, including code files and configuration files that other tools can struggle with.
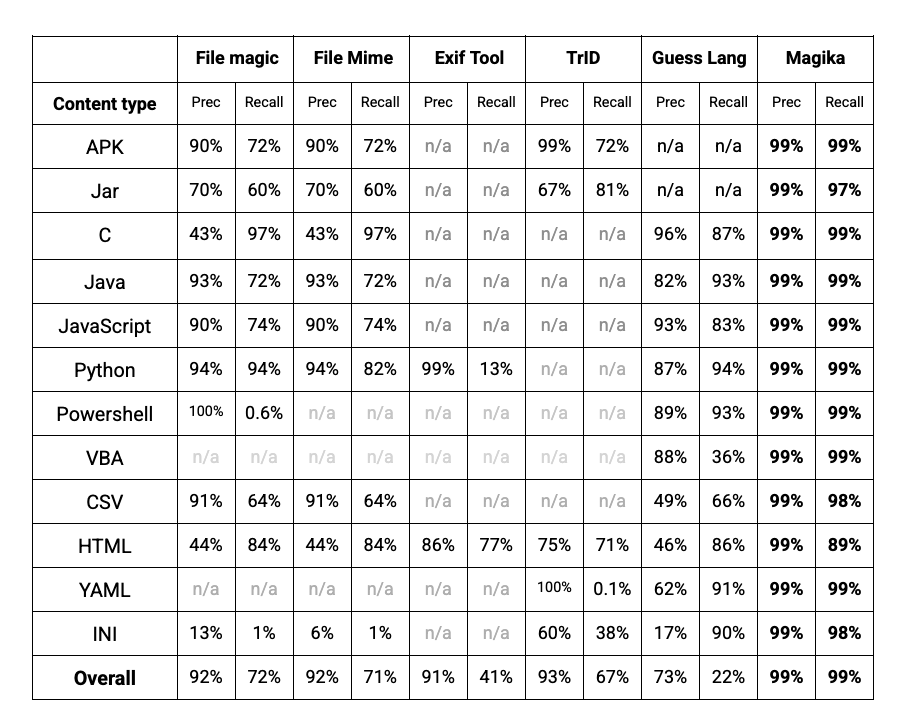 |
| Various file type identification tools performance for a selection of the file types included in our benchmark - n/a indicates the tool doesn’t detect the given file type. |
Magika at Google
Internally, Magika is used at scale to help improve Google users’ safety by routing Gmail, Drive, and Safe Browsing files to the proper security and content policy scanners. Looking at a weekly average of hundreds of billions of files reveals that Magika improves file type identification accuracy by 50% compared to our previous system that relied on handcrafted rules. In particular, this increase in accuracy allows us to scan 11% more files with our specialized malicious AI document scanners and reduce the number of unidentified files to 3%.
The upcoming integration of Magika with VirusTotal will complement the platform's existing Code Insight functionality, which employs Google's generative AI to analyze and detect malicious code. Magika will act as a pre-filter before files are analyzed by Code Insight, improving the platform’s efficiency and accuracy. This integration, due to VirusTotal’s collaborative nature, directly contributes to the global cybersecurity ecosystem, fostering a safer digital environment.
Open Sourcing Magika
By open-sourcing Magika, we aim to help other software improve their file identification accuracy and offer researchers a reliable method for identifying file types at scale.
Magika code and model are freely available starting today in Github under the Apache2 License. Magika can also quickly be installed as a standalone utility and python library via the pypi package manager by simply typing pip install magika with no GPU required. We also have an experimental npm package if you would like to use the TFJS version.
To learn more about how to use it, please refer to Magika documentation site.
Acknowledgements
Magika would not have been possible without the help of many people including: Ange Albertini, Loua Farah, Francois Galilee, Giancarlo Metitieri, Luca Invernizzi, Young Maeng, Alex Petit-Bianco, David Tao, Kurt Thomas, Amanda Walker, and Zhixun Tan.
By Elie Bursztein – Cybersecurity AI Technical and Research Lead and Yanick Fratantonio – Cybersecurity Research Scientist